Town Hall Presentation by Shawn Egan, PhD and CSO, which is recorded here: Click Here
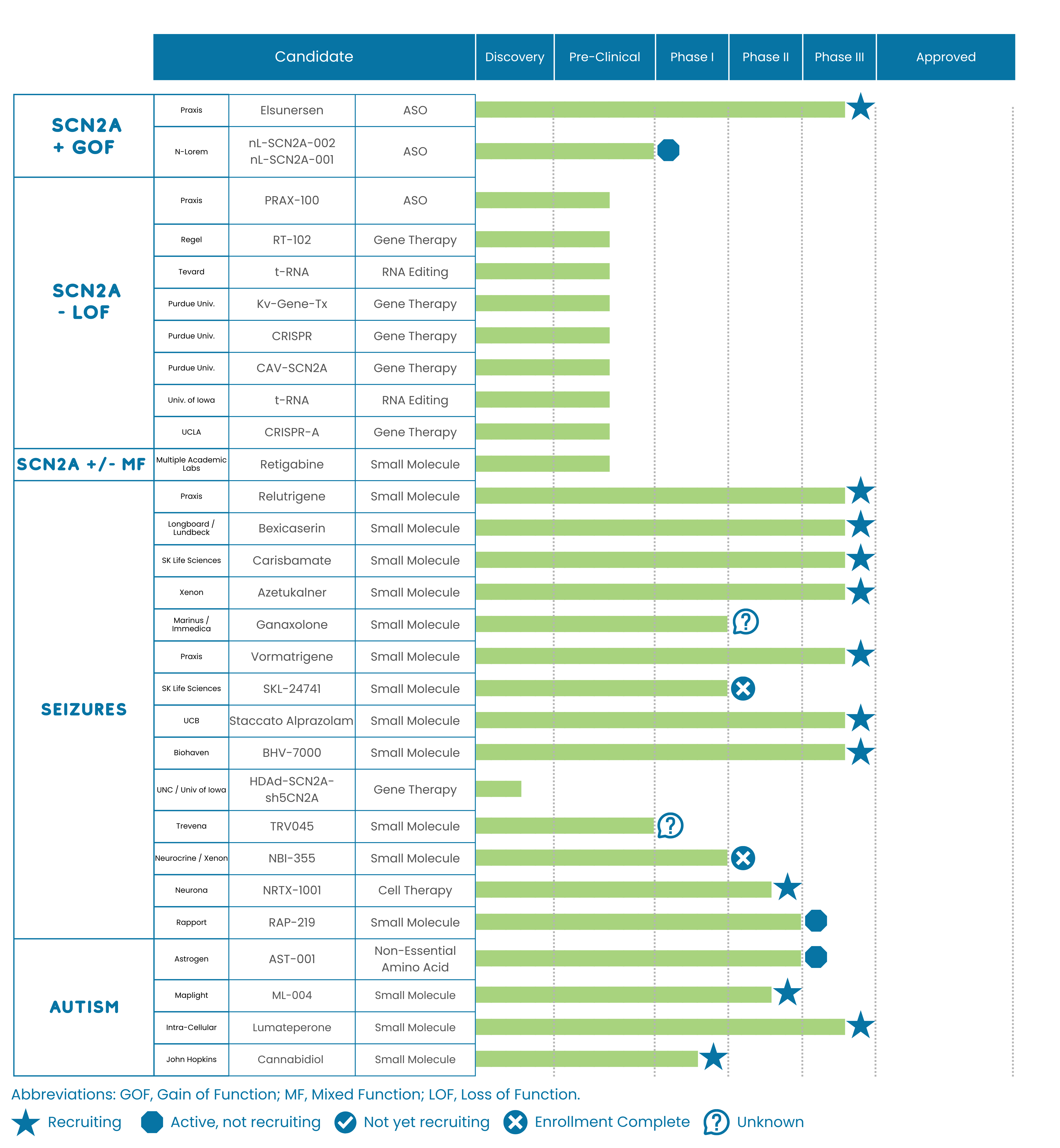
There is excitement in the rare disease community about a cutting-edge treatment that may someday be helpful for other genetic conditions, such as SCN2A-related disorders. In this blog post, we’ll describe this breakthrough and discuss what it means for our community.
On May 15, 2025, The Children’s Hospital of Philadelphia (CHOP) made a historic announcement that Baby KJ was treated successfully with the world’s first personalized gene-editing treatment.
Baby KJ was born with an ultra-rare genetic disorder called CPS1 deficiency, which affects the liver’s ability to process ammonia. Without normally functioning levels of this gene, ammonia accumulates in the blood, leading to toxicity. Toxic ammonia levels lead to severe neurological damage and eventually death within the first weeks to months of life, unless treated with a liver transplant.
In a remarkable race against time, Baby KJ’s team of medical providers, researchers, and industrial partners, were able to design and manufacture a customized treatment in just a few months. The technology that was used to do this is called CRISPR-based gene editing, which can precisely correct disease-causing variants in the human genome. (CRISPR stands for “Clustered Regularly Interspaced Short Palindromic Repeats”—an inconvenient name that simply represents a method that acts like molecular scissor to cut DNA and replace, or “edit,” the wrong DNA letter for the right one.) Once designed and manufactured, the CRISPR gene editing treatment has to be delivered to the right tissue in the body. In Baby KJ’s case, the treatment was encased in lipid nanoparticles that target the liver—the organ that uses the CPS1 gene to convert ammonia into urea so that ammonia can be safely excreted from the body.
Via intravenous infusion, Baby KJ was administered three doses of the CRISPR treatment at ages 7, 8 and 9 months. Following dose 1, Baby KJ’s elevated blood levels of ammonia dropped dramatically. After dose 2, they stabilized to normal levels and Baby KJ is now gaining weight and meeting milestones.
What does this news mean to us?
This is a noteworthy breakthrough for the SCN2A community: For the first time, it demonstrates that a rare pediatric disease can be safely and effectively addressed using the CRISPR system. We are hopeful that the lessons learned from KJ’s treatment can be used for other rare pediatric genetic disorders, including, perhaps, SCN2A.
For this type of treatment to be adapted to children in the SCN2A community, two main challenges remain: (1) Find a way to cross the blood-brain barrier and (2) get regulatory approval for a novel treatment.
First, targeting the liver with lipid nanoparticles for CPS1 is much more straightforward than targeting the brain, as would be needed for SCN2A. Devising vectors and/or other techniques to help CRISPR therapies cross the blood brain barrier efficiently remains a challenge. The sodium channel encoded by the SCN2A gene is widely expressed throughout the brain, and getting the CRISPR treatment to all those cells will require innovative techniques that are currently being developed in labs across the country, but are not yet at the stage that they can be utilized in humans.
The second major challenge is that each different pathogenic variant of SCN2A requires a customized change in the CRISPR treatment, and any change in the CRISPR treatment requires regulatory oversight and approval for administration into humans. The FDA traditionally regards each customized change in CRISPR as a totally new drug. Every new drug requires years of preliminary safety and efficacy data collected from treated animal models before the FDA allows treatment in humans. Yet, there is hope on this front. As recently as June 6, 2025, FDA leadership has expressed willingness to exhibit regulatory flexibility in the context of gene editing treatments for rare disease (https://www.statnews.com/2025/06/06/fda-appears-open-to-relaxed-regulations-on-rare-disease-gene-therapy/).
Baby KJ and his family were brave pioneers that enabled this breakthrough. The many researchers, clinicians, and drug developers involved worked night and day to make medical history, and now, such treatment avenues are more possible for our community. It is certainly a reason to hope. Now it is our responsibility to help educate regulators* of the dire need for gene editing treatments for our community, and continue to encourage industry by demonstrating our willingness to participate in natural history studies and clinical trials.
For more information on Baby KJ’s story, please see the peer-reviewed study published by The New England Journal of Medicine here: https://www.nejm.org/doi/full/10.1056/NEJMoa2504747.
Do you want to help our community advance treatments today? Here’s how:
Participate in the Dragonfly Natural History Study: scn2a.iamrare.org
Donate to the FamilieSCN2A Foundation to support these efforts: scn2a.org/donate.html
If you’re excited to learn more, please attend the FamilieSCN2A Conference this month and our monthly Town Hall Meetings for further research updates.
Do you want to help our community advance treatments today? Here’s how:
Educate regulators about life with SCN2A-related disorders by attending an externally-led Patient Focused Drug Development meeting with the FDA. More on this in a future post!
New Publication: What we can learn from the first personalized CRISPR-treated baby to tackle genetic brain disorders
Author: Yang Yang, PhD
Journal: Neuron (2025)
https://www.scn2a.org/wp-content/uploads/2025/11/Neuron-2025-baby-KJ-to-brain-disorders.pdf